
4. Opinion: Choosing a Molecular Modeling Program
Kristen Procko
The opinions expressed in this perspective are those of the author and do not necessarily reflect the views of BioMolViz.
Introduction
An instructor seeking to incorporate biomolecular visualization instruction into their classroom faces a significant initial hurdle: selecting a program to model macromolecules. It can be challenging—sometimes even frustrating—to learn any new computer program. An instructor needs to identify a modeling program that they are comfortable using, especially if they plan to teach students to use it as well.
In 2021, I was the lead on a manuscript that presented a method for active site modeling using four different programs (Procko et al. 2021). BioMolViz does a significant amount of biomolecular modeling to create figures and structures for our students, as well as images for assessments that probe students’ visual literacy. The team presented four programs for modeling an enzyme active site in that paper, but limited our comparisons to specific facts about interface and usage. I was a self taught PyMOL user at the time, who had learned everything I knew from the PyMOL Wiki, and I was sharing what I’d learned through my YouTube channel. I was fortunate to have team members with expertise in Chimera/ChimeraX, iCn3D, and Jmol, who taught me how to navigate the Graphical User Interfaces (GUIs) of these programs, and helped me write the paper. I added one additional program, Mol* (Molstar), to my repertoire to write this book. In this perspective, I will share what I feel are the highlights and limitations of each program, and I will present how I would choose a program if I were approaching the task anew.
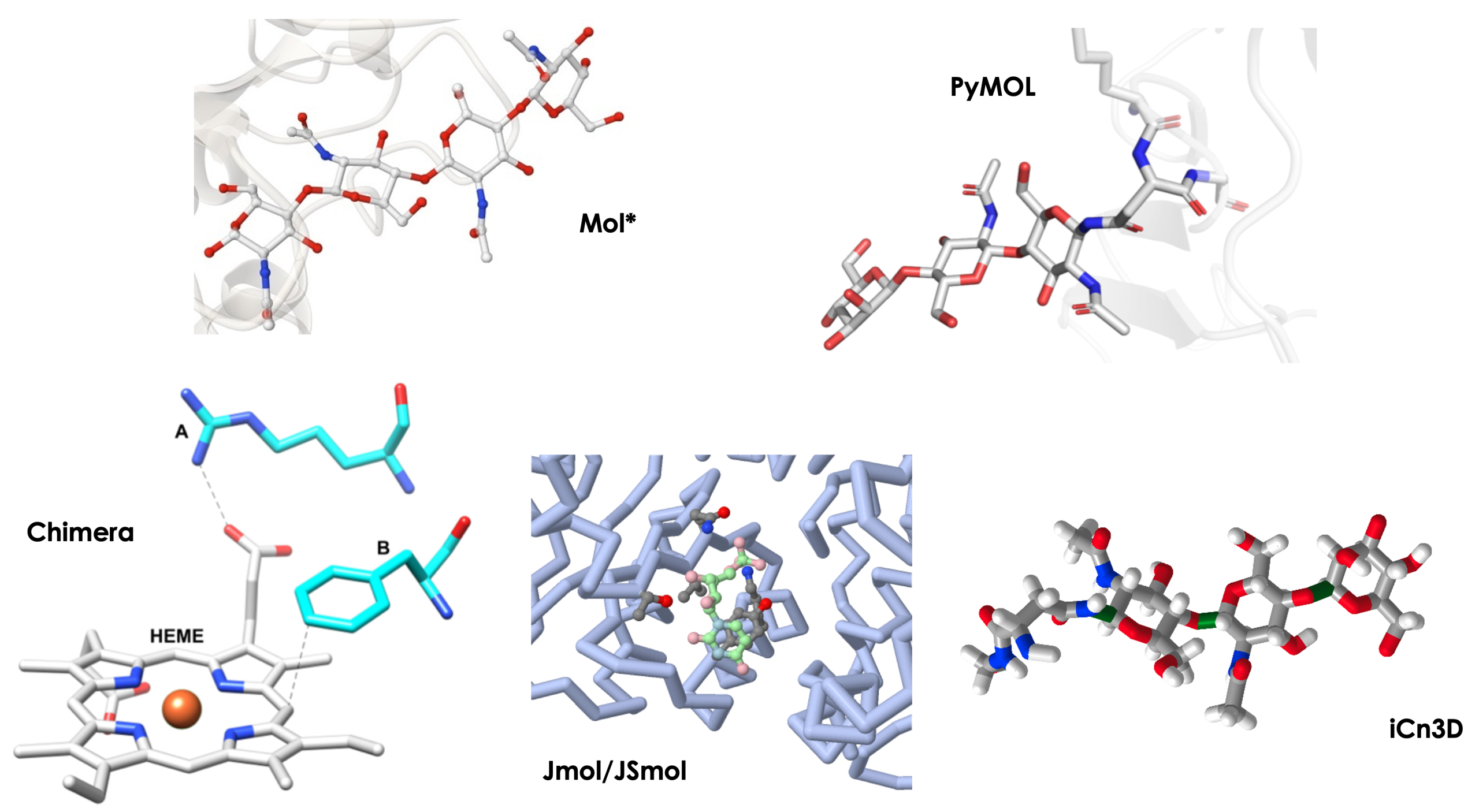
Best Features and Limitations
Summary Table
| Feature / Program | Chimera | iCn3D | Jmol | Mol* | PyMOL |
| Free for educational use | ✓ | ✓ | ✓ | ✓ | ✓ |
| Color by object | ✓ | ✓ | ✓ | ✓ | ✓ |
| Display a surface of a macromolecule | ✓ | ✓ | ✓ | ✓ | ✓ |
| Change the carbon atom color of a selected object easily | ✓ | – | ✓ | ✓ | ✓ |
| Apply a mutation to a nucleic acid or protein structure | ✓ | ✓ | ✓ | – | ✓ |
| No command line needed to unlock full program potential | – | ✓ | – | ✓ | – |
| Heavily used in research | ✓ | – | – | – | ✓ |
| Can produce high resolution images | ✓ | ✓ | ✓ | ✓ | ✓ |
| Essentially infinitely scalable image output; extensive control over output settings | ✓ | – | – | – | ✓ |
Chimera/ChimeraX
Best Features: The visualization in Chimera/ChimeraX is gorgeous. The initial view upon loading a structure is information-rich, and displays some residues near ligands as sticks, so it is easy to spot some interacting atoms right away. The clickable tool icons at the top make the user interface quite intuitive. With knowledge of command-line syntax, a user has a great deal of control over the display. This is a powerful program, which is often stable with very large structure files, making it a top choice for structural biologists working with Cryo-EM structures.
Limitations: Students must download a program to their computer, adding a layer of complexity to getting started. The user should learn some command line syntax to unlock the full potential of the program. The undo feature sometimes didn’t always faithfully reverse the last executed command when I was learning the program, but it does seem to have improved recently.
iCn3D
Best Features: This is a fully online program that runs in a web browser; therefore, the user does not need to download and install a program. There is no need to input typed commands (although advanced users can), so everything the user needs to operate the program is easily found in dropdown menus, which in some cases, open fairly intuitive popup windows. The menu to display molecular interactions is extensive, and, conveniently, different interactions are color coded. The program can load MMDB files, which link 3D structure information with sequence databases and Pubchem, allowing for simple displays of domain and alignment tools, as well as structure/function properties. Possibly the best feature of iCn3D is that it allows the user to generate a link to share an editable session with others—perfect for teaching.
Limitations: In some structures, hydrogen bonds seem to be missing (observed by comparing to the same structure analyzed with PyMOL or ChimeraX). The manual measurement tool can be applied here, but manual measurement in this program is clunky because the user needs to reselect the measurement tool for every measurement. The highest quality picture file output from iCn3D is also of lower quality than outputs from PyMOL and Chimera. Coloring individual atoms of a ligand to differentiate the ligand from displayed protein side chains is very difficult compared to PyMOL and Chimera. Although the share link is a wonderful feature, I once I noticed an older link did not faithfully render the structure as I knew I had from a description I’d written. Also, there are occasionally issues with generating share links when many steps are used to render a structure.
Jmol
Best Features: JSmol is an online version of Jmol that can be run in a browser, and there are several websites that host Jmol interfaces. The Jmol output is a JPEG that can be directly embedded in a document or reopened in Jmol and edited, whereas other programs have a specific file type for working in the program, and the user needs to output a separate image file. The undo button in Jmol is quite robust, and seems to faithfully undo the last executed command every time I tried.
Limitations: The Jmol GUI is limited, necessitating the need to use the command line extensively, and some commands are not well-documented. Because of this, I found Jmol the most challenging program to generate an active site with. Specifically, it is very difficult to automatically generate hydrogen bonds in the program, and we had to find a workaround that would not be intuitive for a beginner.
Mol* (Molstar)
Best Features: Mol* is a web-based program that has been integrated into the RCSB PDB, making it easy to play around with a structure as you look over the rich information available about the structure in the PDB. Clicking on a part of the structure renders nearby amino acid residues, water molecules, etc. even showing color-coded molecular interactions automatically. The menus to render the different molecular components (water molecules, protein, glycans, ligands) are automatically displayed and quite intuitive to use. The save file is very convenient to reload. It can simply be dragged and dropped back into Mol* in a browser window.
Limitations: The program is not capable of mutagenesis, so it doesn’t have a built in feature that lets students explore the change of one amino acid residue for another. Some of the more advanced features require the user to navigate away from the PDB to the web version of Mol*.
PyMOL
Best Features: Selecting residues and ligands is easiest in this program, as selection is executed with a simple click on the structure. Clicking another object adds it to the selection—there are no fancy key combinations as with other programs to add to the selection. Selections appear as a new menu item directly on the GUI, so they are easily accessible to operate on individually, no popup windows or semi-hidden menu items are required to reveal them. The measurement wizard is intuitive, allowing the user to manually measure distances easily and the program creates a new object for each measurement or group of measurements with an intuitive setting change in the popup menu. PyMOL3 has introduced a new scenes menu that shows a thumbnail of the saved scene, and an improved movie-making interface.
Limitations: Students must download a program to their computer, and PyMOL does not work well on some light computers, like a Chromebook. The user should learn command line syntax to unlock the full potential of the program. Previously, the undo feature was not robust—I’m not sure if this is improved yet in PyMOL3. The GUI contains a great deal of menus within buttons, and it is not immediately intuitive that some buttons are actually clickable menus.
My Program Selection
I currently teach biomolecular modeling in my courses to junior-level biochemistry majors. For majors, I think that learning a robust molecular modeling program like PyMOL or Chimera is appropriate, as these programs are often employed in research settings. Jmol is at the bottom of my list, although the Jmol-based interface of Proteopedia is much easier to use (Hodis et al., 2008; Prilusky et al., 2011)
However, if I were introducing molecular modeling to non-majors or to students in their early academic career, I would choose iCn3D or Mol*. Because they are completely browser-based, students do not need to download a program. iCn3D has the additional advantage that a pre-rendered structure can be shared with a simple link. The link could be used to generate a QR code, enabling students to use their tablets or even phones to manipulate the structure. Although I strongly prefer to use all modeling programs on a computer (and PyMOL and ChimeraX are restricted in this way), iCn3D is the rare program that is rather tolerable to use on mobile. This makes iCn3D my favorite choice for ease of use in teaching.
Modeling Skills are Transferrable
Whichever program you choose to learn, you can be confident that the skills you are learning are transferable. As a relatively skilled PyMOL user, I felt very comfortable learning the basics of the four other programs described in this opinion. Simply knowing the basic principles of how to manipulate a molecular model (e.g., changing renderings, measurement) empowered me to play around and find these features in other programs. Executing tasks that were less intuitive in a program did require some web searching and frustration at times. I was fortunate that I could reach out to my colleagues for help, which are the same colleagues that coauthored this book for you, so I hope you will also benefit from my teammates knowledge.
Perusing the first few activities of this book may help you identify an interface you’ll be comfortable with. My main pieces of advice are don’t agonize over program selection, dive in eager to bring some active learning to your classroom, and don’t be afraid to pivot to a new program if your initial selection doesn’t work for you. I hope you have fun learning something new—Happy Modeling!
References
Hodis, E., Prilusky, J., Martz, E., Silman, I., Moult, J., & Sussman, J. L. (2008). Proteopedia-a scientific ‘wiki’ bridging the rift between three-dimensional structure and function of biomacromolecules. Genome biology, 9, 1-10. PMID: 18673581 DOI: http://dx.doi.org/10.1186/gb-2008-9-8-r121
Prilusky, J., Hodis, E., Canner, D., Decatur, W. A., Oberholser, K., Martz, E., … & Sussman, J. L. (2011). Proteopedia: a status report on the collaborative, 3D web-encyclopedia of proteins and other biomolecules. Journal of structural biology, 175(2), 244-252. PMID:21536137 DOI: 10.1016/j.jsb.2011.04.011
Procko, K., Bakheet, S., Beckham, J. T., Franzen, M. A., Jakubowski, H., & Novak, W. R. (2021). Modeling an Enzyme Active Site using Molecular Visualization Freeware. Journal of Visualized Experiments, (178). DOI: 10.3791/63170